喜欢一件事就去做吧,看看究竟是什么,未完待更。
第二章:生物数据库
数据库分类
- 核酸数据库
- 一级核酸数据库,
- 蛋白质数据库
- 一级蛋白质数据库,一级蛋白质结构数据库
- 专用数据库
- 二级核酸数据库,二级蛋白质数据库
文献数据库 PubMed
搜索中:
- Down [AU] 表示搜索的 Down这个人(作者)
- Down [TI] 表示按标题搜索这个词
- Down [AD] 按实验室地址搜索这个词
- Down 在任意地方搜索这个词
一级核酸数据库 GenBank
- NCBI 美国
- ENA 欧洲
- DDBJ 日本
- INSDC 国际核酸序列数据库(由以上三大数据库构成,使得以上的数据库数据都大概相同)
2.4.4 基因组数据库
- ensembl 脊椎动物数据库
- jcvi 微生物数据库
2.6 二级核酸数据库(比较多)
- RefSep 数据库:参考序列数据库,人工精选出的非冗余数据库。
- dbEST 数据库:表达序列标签数据库,不同物种的表达序列标签。
- Gene 数据库:提供基因序列注释和检索服务。
- ncRNAdb :非编码RNA数据库,存放已发表的microRNA序列和注释。
2.7 一级蛋白质数据库
- 一级蛋白质-序列-数据库
- swissprot 人工注释的蛋白质数据库,冗余的小,可信度高。
- RrEMBL 计算机注释的数据库,数据量大,可信度低,冗余量大。
- PIR 蛋白质信息资源数据库,是一个支持基因组学,蛋白质组学和系统生物学研究的综合公共生物信息学资源。
- UniProt 三层数据库
- UniParc:收录所有子库中的蛋白质序列,量大,粗糙。
- UniRef:归纳了UniProt几个主要数据库并将重复序列去除后的数据库。
- UniProtKB:有详细注释并连接了其他数据库,分为:UniProtKB/Swiss-Prot和UniProtKB/TrEMBL。我们主要用Swiss-prot
- 一级蛋白质-结构-数据库
2.8 一级蛋白质数据库PDB-生物大分子3D结构的数据库
- 这些生物大分子除了蛋白质以外还包括核酸及两者的复合物。
2.9 二级蛋白质数据库 Pfam, CATH, SCOP2
- Pfam 蛋白质结构域家族的集合
- CATH
- class
- architecture
- topology
- homologous superfamily
- SCOP2
- class 主要的二级结构成分
- fold 空间的几何关系
- super family 远源的蛋白质进化关系
- family 相近的蛋白质进化关系
2.10 专用数据库 KEGG OMIM
- KEGG 京都基因组百科全书,是关于基因、蛋白质、生化反应及通路的综合生物信息数据库,由多个子库构成。
- OMIM 人类遗传疾病及相关位点的详细信息
第三章:序列比较
3.1序列
- 蛋白质序列:由20个不同字母(氨基酸)排列组成。
- 核酸序列:由4个不同的字母(碱基)排列组成。
- fasta格式:第一行大于号加名称或其他注释,第二行以后每行60个字母(也有80个的,不一定)
比较的目的就是确定相似性
3.3 替换计分矩阵
- 反映残基之间相互替换率的矩阵,它描述了残基两两相似的量化关系。分为:
- DNA替换计分矩阵
- 蛋白质替换计分矩阵。
3种常见的DNA序列的替换计分矩阵
- 等价矩阵
- 转换-颠倒矩阵
- 嘌呤有两个环,嘧啶只有一个环
- 转换环数不变A - G、C - T
- 颠倒环数发生变化,A - C、T - G
- 转换发生的频率要比颠倒高。
- 转换得分为-1 ,而颠倒得分为-5.
- BLAST 矩阵(就是好)
- 令被比对的两个核苷酸相同时得分为+5,反之为-4,
- 这个矩阵广泛地被DNA序列比较采用。
3种常见的蛋白质序列的替换计分矩阵
等价矩阵(较少使用)
- 相同氨基酸匹配得分1,不同氨基酸间替换得分0。
PAM 矩阵(最广泛使用)- 序列的差异度
- 基于进化原理。如果两种氨基酸替换频繁,说明自然界易接受这种替换,那个这对氨基酸得分就高。
BLOSUM 矩阵 - 相似度
关系较远的时候BLOSUM-45更具优势,关系较近的序列用PAM或BLOSUM-62作出的比较差异不大。常用BLOSUM-62矩阵。
3.4 序列两两比较-打点法
3.5 序列两两比较-序列比对法
- needleman-Wunsch 算法,
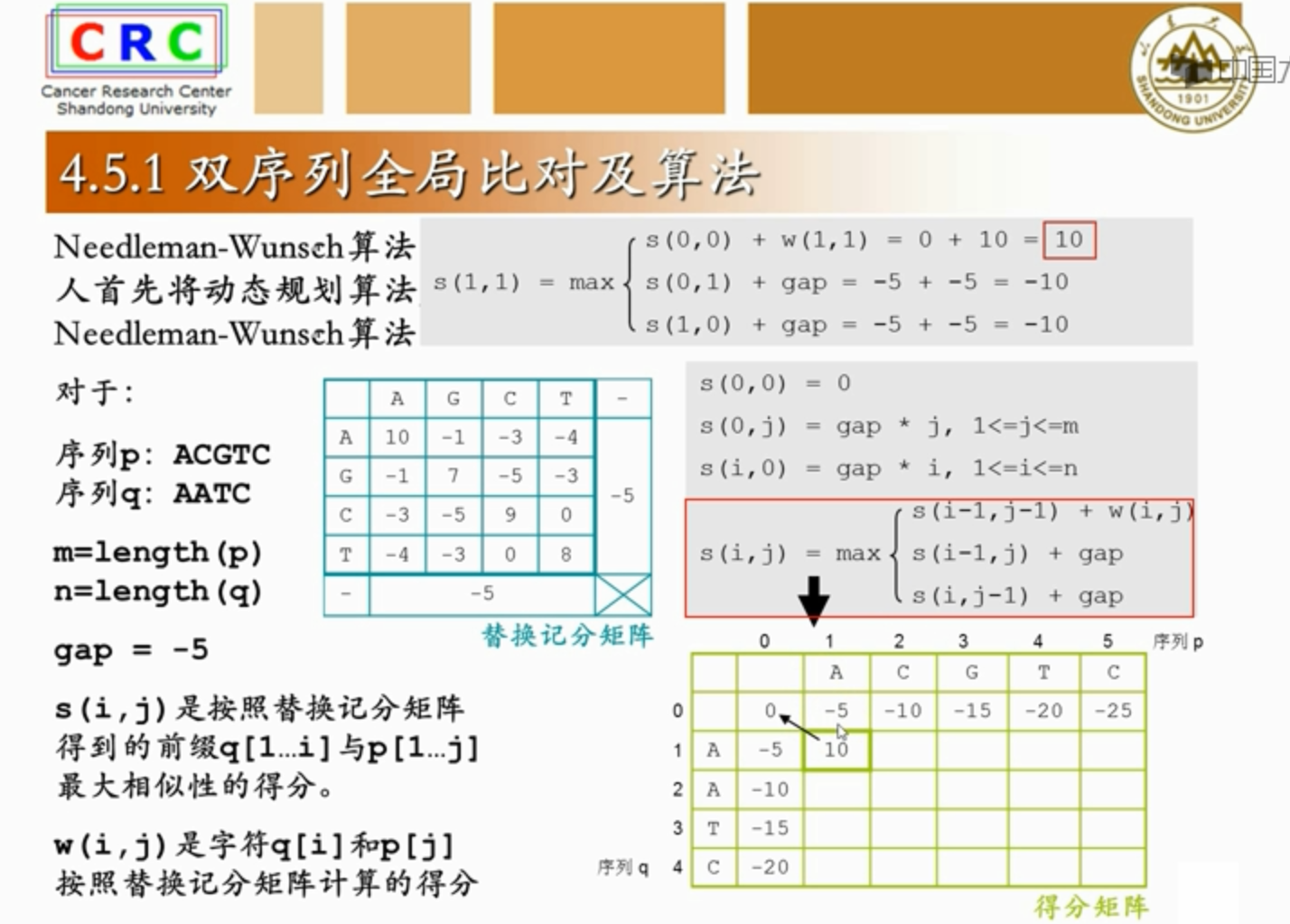
- 全局比对(用于比对两个长度近似的序列)
- 局部比对(用于比对一长一短的两条序列)
3.6 一致度和相似度
- 一致度 = (一致字符串的个数/全局比对的长度)*100%
- 相似度 = (一致及相似的字符的个数/全局比对的长度)*100%
3.7 在线双序列比对工具
- EMBL 全局双序列比对工具
- gap 开头和gap 结尾的意义,
- 设置的gap开头大,比较集中,序列不容易打开,
- gap 开头小,gap 结尾大,序列容易打开,
- EMBL 局部双序列对比工具
比对工具
3.8 BLAST 搜索
- 可以用作寻找相同或相似的序列,一般NCBI、PDB、Uniprot都提供BLAST搜索服务。
- blastp 用蛋白质搜索蛋白质序列的数据库。
- blastn 用核酸序列搜索核酸序列数据库
- blastx 将核酸序列按6条链翻译成蛋白质序列后搜索蛋白质序列数据库
- tblastn 用蛋白质序列搜索核酸序列数据库,数据库中的核酸序列要按6条链翻译成蛋白质序列后再搜索。
- tblastx 将核酸序列按6条链翻译成的蛋白质序列后再搜索核酸序列数据库,数据库中的核酸序列要按6条链翻译成的蛋白质序列后再搜索。
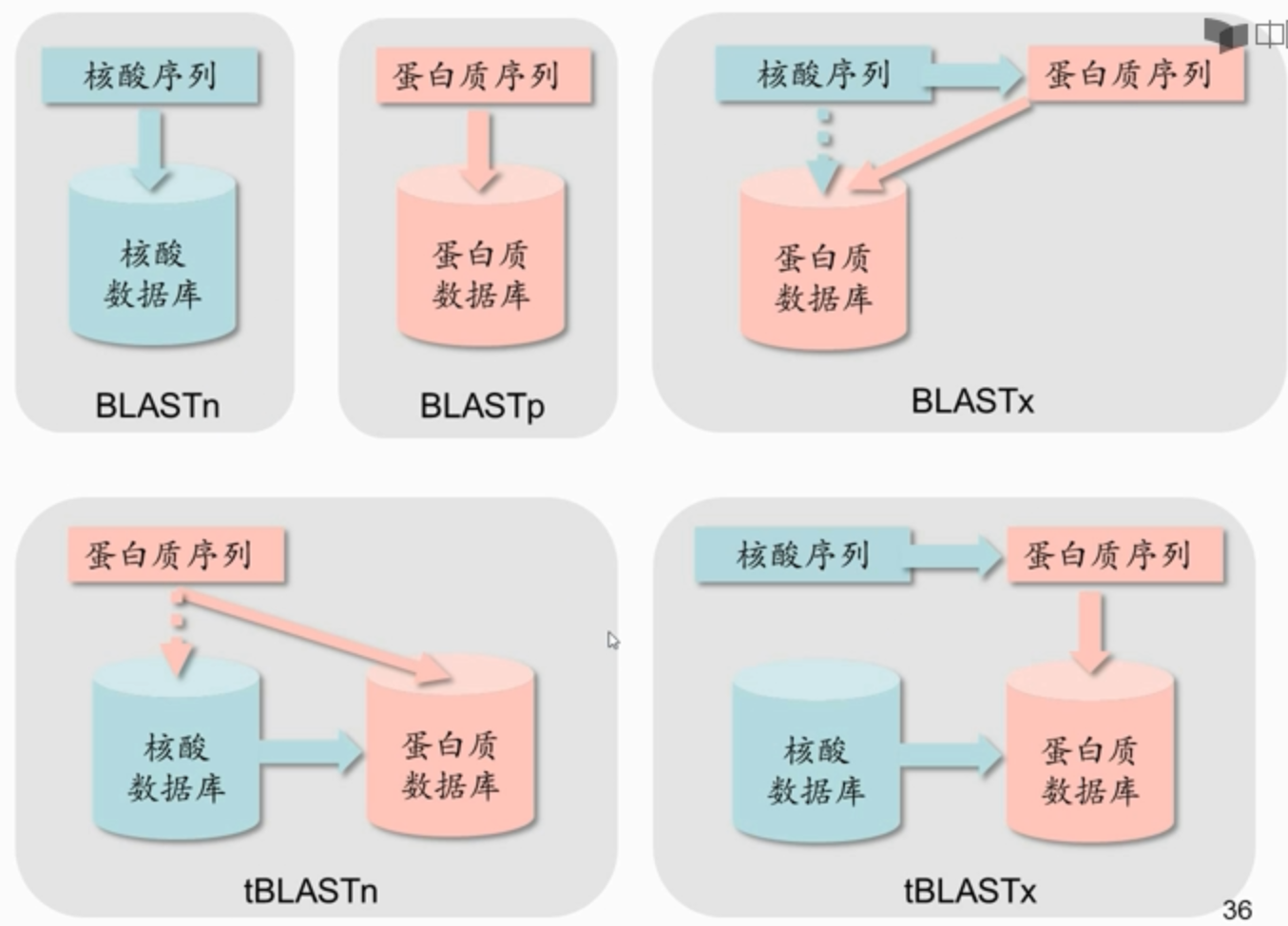
只运用blast只能搜索到十分相近的序列,而其他哪些远亲就找不到了,就是朋友的朋友找不到了。
PSI-BLAST(特异性迭代BLAST) 可将搜索出来的序列再用来搜索。
PHI-BLAST(模式识别BLAST)能找到与输入序列相似的符合某种特定模式的序列。
- 如:以Asn开始,然后紧跟除了Pro之外的任何一个氨基酸,再紧跟Ser或Thr,再紧跟除了Pro外的任何一个氨基酸。写法要用到正则表达N{P}[ST]{P}
“{}”表示除什么以外,”[]”表示其中之一,”x”表示任意字符,”(3,7)”表示3到7个前面的字符。
一般我们可以用smartBLAST
网上的免费的搜索工具

3.9 多序列比对介绍
两条以上的生物序列进行全局比对。
用途:
- 确认:一个未知的序列是否属于某个家族
- 建立:系统发生树,查看物种间或序列间的关系。
- 模式识别
- 已知推未知
- 预测蛋白质/RNA二级结构等,,
使用多序列的几点要求:
- 太多不行。不能超过50条,一般10-15条
- 关系太远不行。
- 关系太近不行。
- 短序列不行。应该都是差不多的序列。
- 有重复序列的不行。
多序列对比工具
3.10 在线序列比对
序列文件格式:
- score_html file 很好看的网页格式
- clustalw_aln file 多序列比对用的格式
- fasta_aln file 一条序列一条序列
- phylip file 方便建树用的格式
格式转换工具:fmtseq sequence conversion
3.11多序列比对的编辑和发布
对多序列比对结果进行彩色显示和手工编辑
- Jalview
- 多序列比对美化工具

3.12寻找保守区域
一个重要的问题:我们在多序列比对中获得什么?
- 答案是:保守区域
weblogo -序列标识图
可用于把重复出现的字母变成某个字母的长度。
MEME - 序列基序
prints 指纹图谱数据库-蛋白质的功能与序列关系
prints 是蛋白质序列指纹图谱数据库,储存了目前已发现的绝大多数蛋白质家族的指纹图谱。对于一个陌生的蛋白质,只要看看它的序列是否符合某个家族的指纹图谱。一个陌生的蛋白质,只要看看它的序列是否符合某个家族的图谱就可以对他进行分类并预测它的功能。
一个蛋白质的指纹就是一组保守的序列基序,用于刻画蛋白质家族的特征。这些基序由多序列比对结果获得,且他们在氨基酸序列上是不相邻的,但是在三维结构中,他们可能紧密结合在一起。
掌握序列比较的方法并学会分析它的结果,就可以把诸多看似零散的信息拼接成一个完整的魔方。
第四章:分子进化与系统发生树
4.1 进化的故事
- 拉马克的用进废退和达尔文的进化论,达尔文的更适合解释生物进化现象。
4.2 基本概念
- 如何研究进化:
- 一、生物化石;
- 二、比较形态学、比较解剖
- 三、分子进化:利用软件,从分子水平上(DNA、RNA、蛋白质序列)构建各种生物间的系统发生树。准确度依赖软件的优劣及参数的设置。
基本概念:
- DNA、RNA或蛋白质序列包含了物种的所有进化史信息;
- 分子钟理论:一个特定蛋白质的进化变异的速度在不同物种中是基本恒定的。即两个蛋白质的序列越相近,他们距离共同祖先就越近。
不同的同源
- 相似的序列来源有两种途径,一种是来源于共同祖先的相似序列,一种是来源于不同祖先的序列。意思就是,相似序列并不一定是同源序列。
- 同源分:
- 直系同源:来源于同一物种的两个物种的相似序列。
- 旁系同源:可能会进化出新的与原来有关的功能。
- 异同源:通过水平基因转移。来源于共生或病毒侵染所产生的相似基因。
不可以说是同源性80%,只能说是相似性80%,会被人看不起。
4.3 系统发生树(分子树)
意义:
- 对于一个未知的基因或蛋白质序列,确定其亲缘关系最近的物种。
- 预测一个新的发现的基因或蛋白质的功能。(相似基因有相似功能)
- 有助于预测一个分子功能的走势。
- 追溯一个基因的起源。
系统发生树的种类
- 有根树:反应了树上基因或蛋白质进化的时间顺序,通过分析有根树的树枝的长度,可以了解不同的基因或蛋白质以什么方式和速度进化。
- 无根树:只反映分类单元之间的距离,而不涉及谁是谁的祖先问题。
- 做有根树需要指定一个外类群,就是你说研究之外的一类群,怎样就可以确定一个共同的祖先,确定一个根。
物种树
- 1998年伍斯构建了一个涵盖了整个生命界的系统树(物种树)十分庞大。
物种树与分子树
- 物种树是基于整个基因组构建的。(比较粗)
- 分子树是基于不同物种里某个基因或蛋白质序列之间的关系构建的。(比较细)
4.4 系统发生树的构建
方法:
- 基于距离的方法(最快,最不精确)
- 最大简约法
- 最大自然法(最优)
- 贝叶斯推断法(最慢,最精确)
软件:
 如果要发表纯生物学的文章,要两种以上的方法得出一个结果才可以通过
如果要发表纯生物学的文章,要两种以上的方法得出一个结果才可以通过
- 基于距离的UPGMA算法
- 非加权分组平均法(UPGMA)
- 用序列间不同的碱基数目作为序列间的距离度量。
- 用什么序列构建序列发生树?
- 如果DNA 序列两两之间的一致度大于70%,就用DNA序列。
- 如果DNA序列两两之间的一致度小于70%的话,DNA序列和蛋白质序列都可以用。(一般用蛋白质序列)
4.5 MEGA7构建系统发生树
免费、支持多操作系统、被业界认可
设置系统发生树的几个设置:
- test of Phylogeny : bootstrap method
- Model/Method : p-distance
- Gaps/Missing Data Treatment :
- Complete deletion(序列间不同残基的个数来作为度量单位的话)
- partial deletion (部分删除,NG方法)
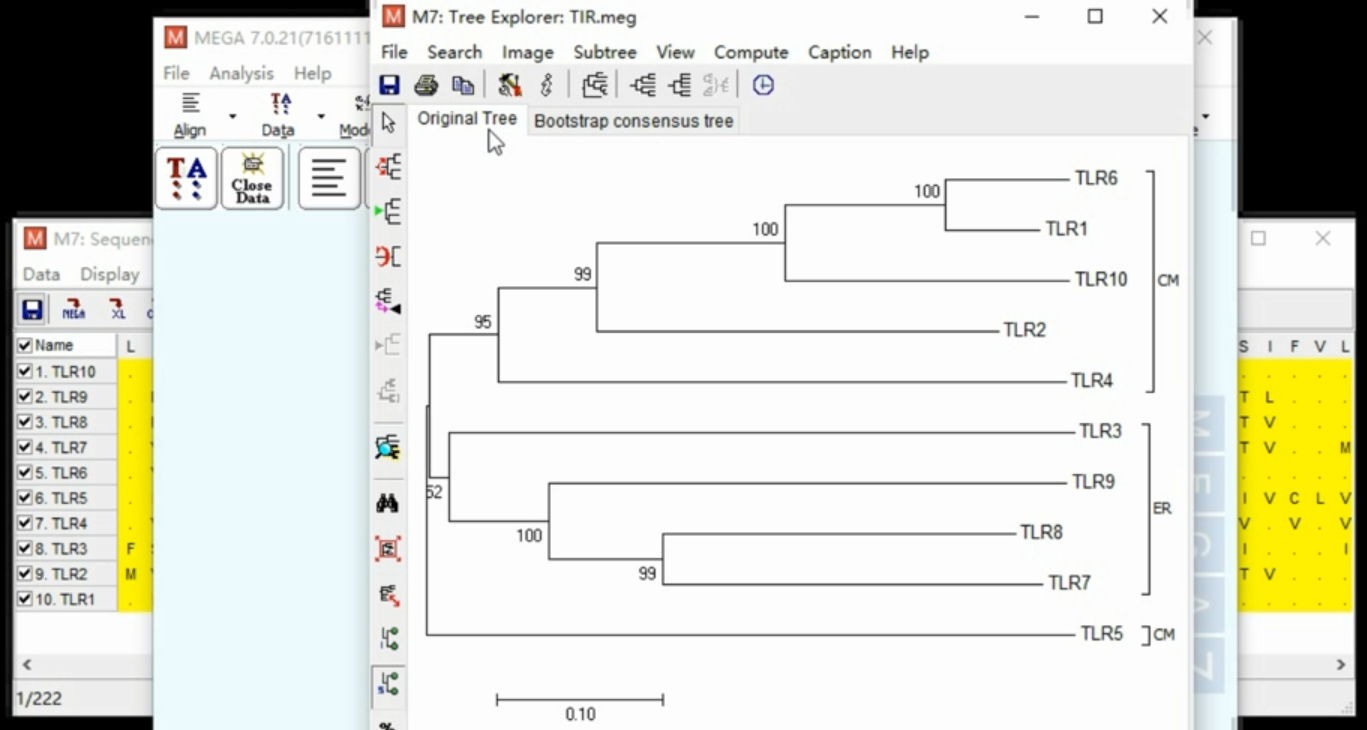 - 树上的数字表示可信度
-
- 树上的数字表示可信度
-
第五章:蛋白质结构的预测与分析
5.1 蛋白质的结构
意义,把蛋白质序列三维化
- 蛋白质的结构
- 一级结构:氨基酸序列
- 二级结构:周期性的结构构象,阿尔法螺旋、β折叠等
- 三级结构:整条多肽链的三维空间结构
- 四级结构:多个亚基形成的复合体结构,如三聚体、四聚体等
5.2 蛋白质的二级结构
- 螺旋:常见的就是阿尔法螺旋。
- β折叠:由β折片平行排列而成。
- β转角:如果肽链发生了急转弯,这个转弯结构叫β转角。
- 无规则卷曲:无规则松山结构。
- 图形中
- E、黄色箭头:β折片结构
- G、波浪线:螺旋结构
- T、小鼓包:转角
- H、代表阿尔法螺旋
- 没有字母的结构就是松散的结构

DSSP(蛋白质二级结构词典)
DSSP并不预测二级结构,而是根据二级结构的定义对已经测定的三级结构的蛋白质的各个位置指认出是哪种二级结构。不能通过氨基酸序列得到DSSP文件(二级结构信息),只能是通过三级结构得到。
DSSP网址
- 一般数据库不提供二级结构的文件(DSSP), PDB中有ss.txt的文件(储存有一级、二级总的结构),可以下载。
蛋白质二级结构的预测
- 对于未知的结构的蛋白质,可以通过氨基酸序列,预测其二级结构。
- 一般邮箱只能是学生邮箱,教育的邮箱,其他的商业邮箱不行。
5.3 蛋白质的三级结构
- 蛋白质结构的测定方法
- X射线衍射法(主要测定方法,能结晶的蛋白质)
- 核磁共振法(不能结晶的蛋白质,测定质量很小)
- 冷冻电子显微镜法(价格高)
- PDB文件是通过存储每一个原子的3D坐标来存储蛋白质的空间结构的。这些坐标可以被三维可视化的软件读取,并展示出来。
- 蛋白质三维可视化软件:
- VMD(与Pymol差点,但免费)
- Maestro
- Pymol(优秀,但收费)
5.4 三维可视化软件 VMD
- 第一节讲了些文件的导入和鼠标的使用。(菜单栏上都有)
- 第二节Graphical(绘图的),里面能更改蛋白质的显示效果_连接
- 第三节multiple representations
5.5 计算方法预测三级结构
实验三高问题:材料要求高、实验仪器造价高、实验耗时高
计算方法:
- 从头计算法
- 同源建模法(首选)
- 穿线法
- 综合法
同源建模法 SWISS-MODLE:
相似的氨基酸序列对应着相似的蛋白质结构
- 找到与目标序列同源的已知结构作为模板(一致度>30%)
- 目标序列与模板序列创建序列比对,通常软件自动创建的序列比对还要工校对。
- 根据第二步创建的序列比对,用同源软件预测结果模型。
- 评估模型质量,并根据评估结果重复以上过程,直到模型质量合格。

